Podczas gdy firmy farmaceutyczne skupiają się na poszukiwaniu leków przeciwko gorączce krwotocznej Ebola, istotnej wskazówki zdaje się nam dostarczać uszkadzające mózg rzadkie schorzenie genetyczne typowe dla populacji Żydów aszkenazyjskich. Pacjenci cierpiący na podtyp C1 choroby Niemanna-Picka nie chorują na gorączkę Ebola, wzbogacając tym samym listę par schorzeń wykształconą wskutek fascynującej formy doboru naturalnego.
Polimorfizm zrównoważony, w tej postaci znany też jako przewaga heterozygot (naddominacja) jest świetną ilustracją przebiegu ewolucji. Wspiera też organizm człowieka w walce z najrozmaitszymi intruzami – prionami, wirusami, bakteriami, pierwotniakami czy grzybami.
Podręcznikowym przykładem polimorfizmu zrównoważonego jest ochrona przed malarią, którą zapewnia bycie nosicielem anemii sierpowatej – zjawisko opisane już w 1954 roku.
Krew chorego z w pełni rozwiniętą anemią sierpowatą jest zbyt „gęsta”, a same krwinki zbyt zniekształcone, by zapewnić korzystne warunki zarodźcom malarii. Nosiciel ma krwinki zmienione w stopniu wystarczającym, by utrudnić życie pasożytowi, nie na tyle jednak, by utrudnić krążenie krwi czy wywołać dolegliwości bólowe i niedokrwistość typowe dla pełnego spektrum choroby. Alfa-talasemia, hemoglobinopatia HbC czy fawizm to inne choroby jednogenowe zmieniające czerwone krwinki w sposób zmniejszający podatność na malarię.
Powiązań pomiędzy wspomnianym na początku schorzeniem genetycznym a gorączką krwotoczną Ebola zaczęto się doszukiwać w roku 2011, gdy dr Thijn Brummelkamp wraz zespołem (Whitehead Institute for Biomedical Research) zaraportowali wNature, że komórki pacjentów z rzadką jednogenową chorobą (z podtypem C1 choroby Niemanna-Picka właśnie) wykazują odporność na infekcję wirusem Ebola.
Zmutowany w tej chorobie gen koduje białko transportowe w normalnych warunkach wiążące cholesterol, będące też jednak receptorem dla wirusa. Nosiciele z o połowę mniejszą niż zwykła liczbą receptorów być może mogliby być częściowo chronieni przed gorączką krwotoczną (sytuację nosicieli rozważa niedawny artykuł w Wall Street Journal, który nie cytuje jednak klasycznego przykładu anemii sierpowatej – przypomnieli o nim komentatorzy).
Oczywiście powyższe przykłady, w których jedno schorzenie zdaje się wykluczać drugie, są opcją bardzo uproszczoną – relacje pomiędzy nami i naszymi patogenami bywają dużo bardziej skomplikowane. Tak jak i nie zawsze korzystny efekt niektórych mutacji wymaga drugiego, infekcyjnego, gracza, by dać się zaobserwować. Niska krzepliwość hemofilityków chroni ich przecież przed skutkami niebezpiecznej nadkrzepliwości, a drastycznie niski poziom cholesterolu w surowicy cierpiących na zespół Smitha-Lemliego-Opitza przyczyniający się też do niepełnosprawności intelektualnej i licznych wad wrodzonych chroni przed chorobami układu sercowo-naczyniowego.
Przykłady polimorfizmu zrównoważonego to prawdziwe historie detektywistyczne, w których pierwszy krok polega na zastanowieniu się co właściwie sprawia, że na pierwszy rzut oka szkodliwy gen recesywny nie został dotąd wyeliminowany i wciąż krąży w populacji. Odpowiedzią może być nowość danej mutacji, jednak dużo częstszym rozwiązaniem zagadki będzie ochrona dostarczana przez przewagę heterozygot. Oto niektóre spośród podobnych opowieści.
MUKOWISCYDOZA I BIEGUNKA
U chorego na cholerę toksyny bakteryjne otwierają błonowe kanały chlorkowe w komórkach nabłonkowych jelita cienkiego, przez co woda wraz z elektrolitami wypływa strumieniem biegunki. Uszkodzone w mukowiscydozie budujące kanały chlorkowe właśnie białko CFTR przyczynia się do zjawiska dokładnie odwrotnego – nie mogąc dotrzeć do powierzchni komórki i utworzyć prawidłowej struktury, zatrzymuje w komórce wodę z elektrolitami, zmniejsza ilość wydzielin, wysusza je. Chory z nieprawidłowym białkiem CFTR nie zachoruje na cholerę, podczas gdy nosiciel z wystarczającą liczbą funkcjonalnych kanałów chlorkowych, by sprawnie oddychać, ale zbyt małą, by oddać pole bakteriom i ich toksynom, nie będzie musiał znosić objawów żadnej z chorób.
Epidemie cholery przetaczające się poprzez historię ludzkości mogły się przyczynić do faworyzowania czy to osobników cierpiących na mukowiscydozę, czy też samych nosicieli. Geografia i historia zresztą sugerują, że opowieść sięga dużo dalej w przeszłość, jako że początków mukowiscydozy doszukuje się w zachodniej Europie, a cholery – w Afryce. Być może inne schorzenie wiążące się z biegunkami – dur brzuszny – przyczyniło się pierwotnie do przewagi zmutowanych alleli.
Bakterie odpowiedzialne za dur brzuszny, Salmonella typhi, wnikają do komórek nabłonka jelitowego właśnie przez opisane wyżej kanały. Gdy te nie dotrą do powierzchni komórki, bakterie nie zdołają się dostać do środka. W przypadku komórek nosicieli dostanie się ich do wnętrza niewiele. Ochrona przed infekcjami biegunkowymi może zatem być właśnie tym, co sprawiło, że mukowiscydoza wciąż „krąży”. [przyp. tłum. pojawiają się także sugestie, że za część presji selekcyjnej faworyzującej nosicieli mukowiscydozy może potencjalnie odpowiadać także zmniejszona podatność na gruźlicę]
FENYLOKETONURIA I MYKOTOKSYNY
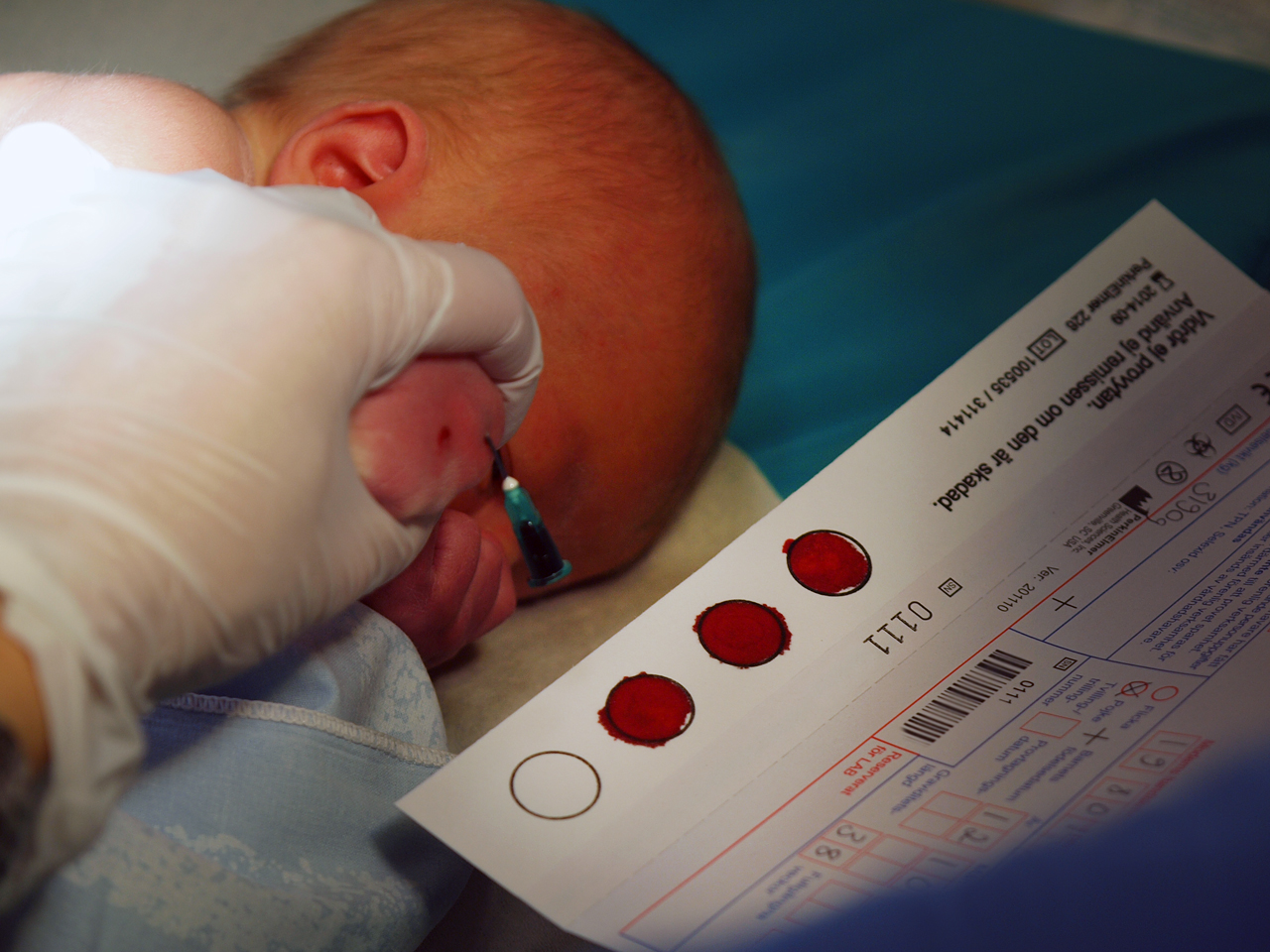
Badania w kierunku fenyloketonurii (PKU) wprowadziły nas w nową erę, erę badań przesiewowych noworodków, przy pomocy opracowanego w 1957 roku testu Guthriego, jednocześnie zaś wiążą się z jedną z moich ulubionych opowieści genetycznych. Badaniu poddaje się krew pobraną z pięty noworodka, by później, pilnując jego diety, móc zapobiec typowemu dla choroby ciężkiemu upośledzeniu rozwoju umysłowego.
Fenyloketonuria to klasyczny przykład wrodzonego błędu metabolicznego. Brak pewnego enzymu przyczynia się do gromadzenia w organizmie jednego z aminokwasów, fenyloalaniny. Kumulacja tejże uszkadza układ nerwowy. Nadmiar fenyloalaniny obserwuje się też u nosicieli wadliwego genu – nie jest on wystarczający, by upośledzać rozwój mózgowia, wystarczy jednak, by chronić przed wytwarzaną przez grzyby toksyną (ochratoksyną A) o działaniu poronnym.
W roku 1986 L. I. Woolf, autorytet na polu fenyloketonurii, opublikował krótkie doniesienie w American Journal of Human Genetics, w którym to tłumaczył w jaki sposób grzyby mogły pomóc utrzymać się w populacji rzadkiej chorobie dziedzicznej. Ochratoksyna A będąca pochodną fenyloalaniny może wiązać się z enzymem umieszczającym ten aminokwas w odpowiednim miejscu łańcucha białkowego. Uniemożliwiając w ten sposób włączanie fenyloalaniny do budowanych białek, powoduje szybkie zatrzymanie rozwoju zarodkowego. Chyba że organizm ciężarnej dysponuje nadmiarem tego aminokwasu, co odwraca działanie toksyny. To właśnie ma miejsce u nosicieli PKU. Dzięki badaniom przesiewowym fenyloketonuria jest obecnie diagnozowana wystarczająco wcześnie, by wdrożyć odpowiednie postępowanie, ochratoksyna A tymczasem w Europie Wschodniej przyczynia się do rozwoju ciężkiej choroby nerek zwanej nefropatią bałkańską.
Nieprzypadkowo fenyloketonuria jest szczególnie częsta w Szkocji i Irlandii. W wilgotnym klimacie tych rejonów wytwarzające ochratoksynę grzyby z rodzajów Aspergillus i Penicillium szczególnie chętnie rosną na przechowywanym ziarnie, które niezależnie od jego stanu spożywali przymuszeni głodem mieszkańcy. Jako że ciężarne nosicielki miały większe szanse na donoszenie zdrowych dzieci niż kobiety bez obciążeń genetycznych, które częściej roniły wskutek działania mykotoksyny, częstość mutacji w populacji zwiększała się. Tak właśnie działa dobór naturalny. Ewolucja.
PRIONY I KANIBALIZM
Priony to białka zdolne do zwijania się w postacie infekcyjne przenoszące tzw. pasażowalne encefalopatie gąbczaste. Najbardziej znane spośród nich są związane ze spożyciem nieszczęsnych białek, jak chociażby choroba wściekłych/szalonych krów (gąbczasta encefalopatia bydła). Klasycznym przykładem jest kuru, niszcząca struktury mózgowe choroba, występująca u członków plemienia Fore (zwłaszcza kobiet i dzieci) z Papui Nowej Gwinei, którym zdarzało się spożywać mózgi czcigodnych zmarłych krewnych. Rząd Australii ukrócił te praktyki w połowie lat pięćdziesiątych.
Jesteśmy zresztą zdolni do wytwarzania swoich własnych białek prionowych – gen kodujący je znajduje się na chromosomie 20. W normalnych warunkach występuje ono w mózgu dość obficie i prawdopodobnie jest istotne dla plastyczności synaptycznej we wczesnym rozwoju mózgu. Opowieść jest tu nieco bardziej skomplikowana niż przy Eboli, mukowiscydozie czy fenyloketonurii, ponieważ to samo białko może pochodzić zarówno z zewnątrz, jak i z wewnątrz naszego organizmu, i to samo białko również, w zmienionej postaci, jest tu patogenem. Jest Zmiennokształtnym potrafiącym przybrać formę infekcyjną w samym środku naszych organizmów.
Wróćmy na Nową Gwineę. Niektóre kanibalki z plemienia Fore nadal żyją, a badania białek prionowych u 30 spośród nich wykazały, że 23 to heterozygoty pod względem genu dla białka prionowego na chromosomie 20, czyli że posiadają po dwie nieco różniące się wersje tego genu. Tymczasem statystyki, zgodnie z zasadami genetyki populacyjnej, sugerowałyby, że taka sytuacja powinna wystąpić jedynie u 15 z nich.
U nosicielek na jednym chromosomie 20 w genie na pozycji 129 znajduje się sekwencja kodująca aminokwas walinę, a na drugim – metioninę. Taka kombinacja okazuje się w jakiś sposób zapobiegać zwijaniu się białka w formę infekcyjną. Dodam tylko, że wszyscy obywatele Wielkiej Brytanii, u których rozwinęła się choroba szalonych krów mieli na pozycji 129 jedynie metioninę. I tak kanibalizm mógł się przyczynić do nadreprezentacji infekcyjnych form białka prionowego. Chronionych nosicieli powoli w populacji przybywało.
DLACZEGO TO WAŻNE
Przez wiele lat na polu genetyki funkcjonował jeden centralny dogmat – że DNA koduje RNA, które z kolei koduje białka. Zakładaliśmy, że nasze genomy kodują białka i nic więcej zasadniczo nie robią, jakkolwiek, owszem, wspominało się też mgliście o funkcjach kontrolnych. Introny (części genów, które nie kodują białek) „pojawiły się” w 1977 roku i wywróciły do góry nogami naszą uproszczoną wizję. Tak naprawdę większość z nas, zajmujących się genetyką, nigdy nie uważała tej „reszty” genomu za „śmieciowe DNA”. Jeśli dobrze pamiętam, to niefortunne sformułowanie Francisa Cricka, które media natychmiast przejęły i które potem jakoś przylgnęło do tematu.
Dziś wiemy, że genom dysponuje pełnym pakietem najrozmaitszych mechanizmów kontrolnych. Niektóre z nich rzeczywiście wbudowane są w sekwencje niekodujące stanowiące wszak większość genomu, inne tworzą krótkie sekwencje RNA potrafiące zwijać się w formy zdolne do pochwycenia i wyłączenia poszczególnych genów, jeszcze inne tkwią w rozsianych po całym genomie krótkich powtarzających się sekwencjach.
Polimorfizm zrównoważony to kolejna postać informacji genetycznej. Ujawnia pewną opowieść, pewien podtekst, drugie dno naszych genów zapisane przez historię naszych własnych działań i przez plagi, które doprowadziły do upadku niektórych spośród naszych przodków, jednocześnie umożliwiając osobnikom przenoszącym pewne mutacje przeżycie, tak by rozmnażając się, mogli przenoszone sekwencje zachować na przyszłość.
Zrozumienie tych fascynujących par schorzeń genetycznych i zakaźnych może przynieść nowe sposoby walki z infekcjami. Miejmy nadzieję, że pomoże też w walce z wirusem Ebola.
[Część powyższego tekstu bazuje na fragmentach z mojego podręcznika Human Genetics: Concepts and Applications (Amazon nie uwzględnia jeszcze nowego, jedenastego, wydania). Przy każdej kolejnej edycji muszę sama ze sobą walczyć, by ograniczyć się do 2 jedynie przykładów polimorfizmu zrównoważonego – naprawdę trudno wybrać!]
Dostrzegam w naszym kraju pewną tendencję do zamierania dyskusji i polemiki między reprezentantami równych ideologii politycznych. Jako liberał i eks-konserwatysta jestem tym faktem dość zaniepokojony,…
Ponieważ w naszym kraju trwa zalew prawicowych ksiąg „mądrości” politycznej, i nikt z normalnych liberałów (a nie konserwatywnych wolnorynkowców, którzy mienią się być liberałami), nie…
Cenzura istniała zawsze i chyba zawsze istnieć będzie. Niezależnie od tego czy jest ona rządowa (np. wydawnicza prewencyjna lub represyjna), czy obyczajowa (tabu, tyrania opinii).…
Dziennikarka naukowa po doktoracie z genetyki, autorka licznych podręczników i artykułów, zajmuje się też poradnictwem genetycznym dla przyszłych rodziców i prowadzi kursy z genetyki dla studentów Alden March Bioethics Institute of Albany Medical Centerhttp://www.rickilewis.com/
One Reply to “Gdy mutację przeciwstawić infekcji – od anemii sierpowatej do Eboli”
Swietny tekst i doskonale tlumaczenie! Sporo sie nowego dowiedzialam – o anemii sierpowatej wie kazdy student biologii, ale pozostale historie sa rownie a nawet bardziej fascynujace 🙂

 Podczas gdy firmy farmaceutyczne skupiają się na poszukiwaniu leków przeciwko gorączce krwotocznej Ebola, istotnej wskazówki zdaje się nam dostarczać uszkadzające mózg rzadkie schorzenie genetyczne typowe dla populacji Żydów aszkenazyjskich. Pacjenci cierpiący na podtyp C1 choroby Niemanna-Picka nie chorują na gorączkę Ebola, wzbogacając tym samym listę par schorzeń wykształconą wskutek fascynującej formy doboru naturalnego.
Podczas gdy firmy farmaceutyczne skupiają się na poszukiwaniu leków przeciwko gorączce krwotocznej Ebola, istotnej wskazówki zdaje się nam dostarczać uszkadzające mózg rzadkie schorzenie genetyczne typowe dla populacji Żydów aszkenazyjskich. Pacjenci cierpiący na podtyp C1 choroby Niemanna-Picka nie chorują na gorączkę Ebola, wzbogacając tym samym listę par schorzeń wykształconą wskutek fascynującej formy doboru naturalnego.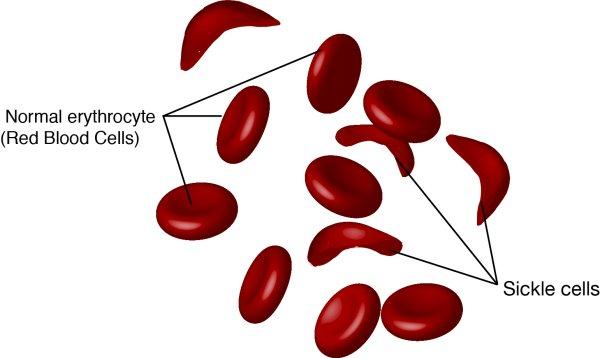 utrudnić życie pasożytowi, nie na tyle jednak, by utrudnić krążenie krwi czy wywołać dolegliwości bólowe i niedokrwistość typowe dla pełnego spektrum choroby. Alfa-talasemia, hemoglobinopatia HbC czy fawizm to inne choroby jednogenowe zmieniające czerwone krwinki w sposób zmniejszający podatność na malarię.
utrudnić życie pasożytowi, nie na tyle jednak, by utrudnić krążenie krwi czy wywołać dolegliwości bólowe i niedokrwistość typowe dla pełnego spektrum choroby. Alfa-talasemia, hemoglobinopatia HbC czy fawizm to inne choroby jednogenowe zmieniające czerwone krwinki w sposób zmniejszający podatność na malarię.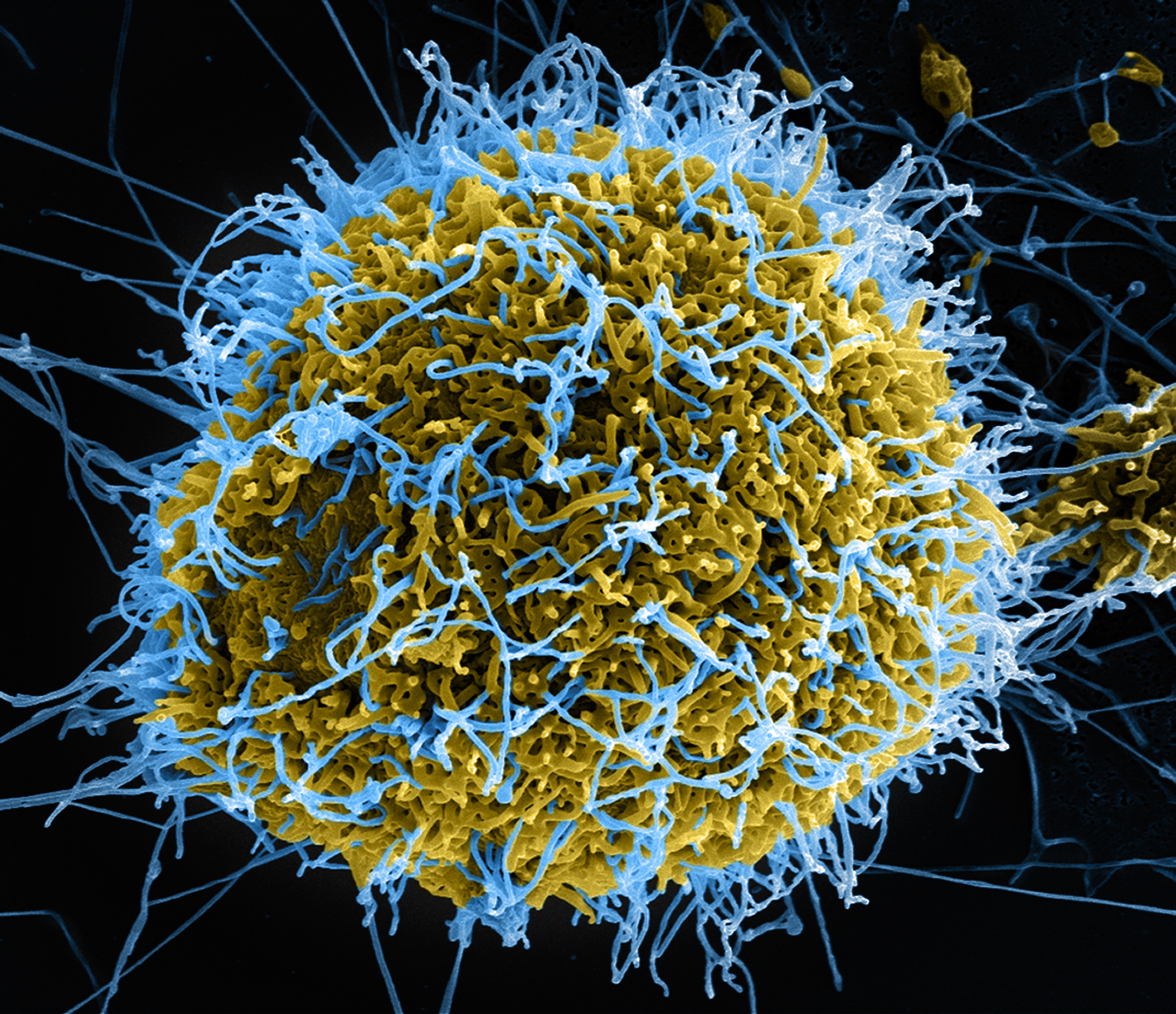 Zmutowany w tej chorobie gen koduje białko transportowe w normalnych warunkach wiążące cholesterol, będące też jednak receptorem dla wirusa. Nosiciele z o połowę mniejszą niż zwykła liczbą receptorów być może mogliby być częściowo chronieni przed gorączką krwotoczną (sytuację nosicieli rozważa niedawny artykuł w Wall Street Journal, który nie cytuje jednak klasycznego przykładu anemii sierpowatej – przypomnieli o nim komentatorzy).
Zmutowany w tej chorobie gen koduje białko transportowe w normalnych warunkach wiążące cholesterol, będące też jednak receptorem dla wirusa. Nosiciele z o połowę mniejszą niż zwykła liczbą receptorów być może mogliby być częściowo chronieni przed gorączką krwotoczną (sytuację nosicieli rozważa niedawny artykuł w Wall Street Journal, który nie cytuje jednak klasycznego przykładu anemii sierpowatej – przypomnieli o nim komentatorzy).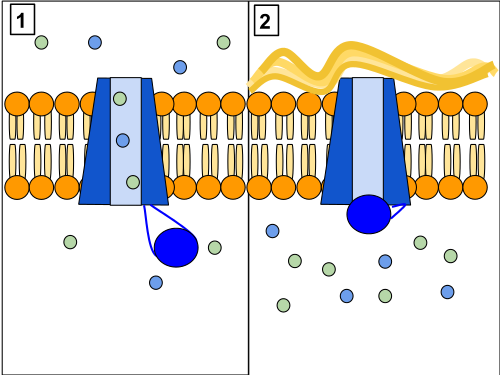
 FENYLOKETONURIA I MYKOTOKSYNY
FENYLOKETONURIA I MYKOTOKSYNY jaki sposób grzyby mogły pomóc utrzymać się w populacji rzadkiej chorobie dziedzicznej. Ochratoksyna A będąca pochodną fenyloalaniny może wiązać się z enzymem umieszczającym ten aminokwas w odpowiednim miejscu łańcucha białkowego. Uniemożliwiając w ten sposób włączanie fenyloalaniny do budowanych białek, powoduje szybkie zatrzymanie rozwoju zarodkowego. Chyba że organizm ciężarnej dysponuje nadmiarem tego aminokwasu, co odwraca działanie toksyny. To właśnie ma miejsce u nosicieli PKU. Dzięki badaniom przesiewowym fenyloketonuria jest obecnie diagnozowana wystarczająco wcześnie, by wdrożyć odpowiednie postępowanie, ochratoksyna A tymczasem w Europie Wschodniej przyczynia się do rozwoju ciężkiej choroby nerek zwanej nefropatią bałkańską.
jaki sposób grzyby mogły pomóc utrzymać się w populacji rzadkiej chorobie dziedzicznej. Ochratoksyna A będąca pochodną fenyloalaniny może wiązać się z enzymem umieszczającym ten aminokwas w odpowiednim miejscu łańcucha białkowego. Uniemożliwiając w ten sposób włączanie fenyloalaniny do budowanych białek, powoduje szybkie zatrzymanie rozwoju zarodkowego. Chyba że organizm ciężarnej dysponuje nadmiarem tego aminokwasu, co odwraca działanie toksyny. To właśnie ma miejsce u nosicieli PKU. Dzięki badaniom przesiewowym fenyloketonuria jest obecnie diagnozowana wystarczająco wcześnie, by wdrożyć odpowiednie postępowanie, ochratoksyna A tymczasem w Europie Wschodniej przyczynia się do rozwoju ciężkiej choroby nerek zwanej nefropatią bałkańską. Priony to białka zdolne do zwijania się w postacie infekcyjne przenoszące tzw. pasażowalne encefalopatie gąbczaste. Najbardziej znane spośród nich są związane ze spożyciem nieszczęsnych białek, jak chociażby choroba wściekłych/szalonych krów (gąbczasta encefalopatia bydła). Klasycznym przykładem jest kuru, niszcząca struktury mózgowe choroba, występująca u członków plemienia Fore (zwłaszcza kobiet i dzieci) z Papui Nowej Gwinei, którym zdarzało się spożywać mózgi czcigodnych zmarłych krewnych. Rząd Australii ukrócił te praktyki w połowie lat pięćdziesiątych.
Priony to białka zdolne do zwijania się w postacie infekcyjne przenoszące tzw. pasażowalne encefalopatie gąbczaste. Najbardziej znane spośród nich są związane ze spożyciem nieszczęsnych białek, jak chociażby choroba wściekłych/szalonych krów (gąbczasta encefalopatia bydła). Klasycznym przykładem jest kuru, niszcząca struktury mózgowe choroba, występująca u członków plemienia Fore (zwłaszcza kobiet i dzieci) z Papui Nowej Gwinei, którym zdarzało się spożywać mózgi czcigodnych zmarłych krewnych. Rząd Australii ukrócił te praktyki w połowie lat pięćdziesiątych.




Swietny tekst i doskonale tlumaczenie! Sporo sie nowego dowiedzialam – o anemii sierpowatej wie kazdy student biologii, ale pozostale historie sa rownie a nawet bardziej fascynujace 🙂